Hemophilia: Everything you need to know!
Published Apr 17, 2021 • Updated Apr 19, 2021 • By Aurélien De Biagi
Mainly affecting men because of the way it is inherited, hemophilia exists in two forms: type A and type B. The former is much more common with approximately 320,000 people affected worldwide.
What is hemophilia exactly? What causes it? How is it treated? What research is being done?
Read on to learn more!

Hemophilia: definition
Hemophilia is a severe hereditary genetic bleeding disorder. There are two types of hemophilia: type A and type B.
Contrary to the image that is often linked to it, people with hemophilia do not bleed more than people without hemophilia.
In fact, a patient living with hemophilia has insufficient clotting factor proteins in the blood (or in some cases, none). In hemophilia A, there are too low levels of clotting factor VIII and in hemophilia B there is a deficiency of clotting factor IX.
Blood coagulation, or clotting, is a complex process involving a number of proteins: the clotting factors. These factors are linked together in a sort of cascade reaction. The 13 factors are therefore necessary for proper clotting. This is why, in the event of a dysfunction or factor deficiency, the blood does not coagulate and bleeding continues. The consequences can be dramatic, with significant bleeding in the event of injury or even spontaneous bleeding in the joints or vital organs.
Repeated bleeding in the joints can even lead to hemarthrosis (blood effusion into joint spaces). This complication will lead to hemophilic arthropathy: permanent joint disease caused by long-term hemarthrosis where the cartilage and later the underlying bone is destroyed over time, causing chronic pain, joint deformity and stiffness. Damage to a vital organ such as the brain can have fatal consequences.
Whatever the type of hemophilia (A or B), the cause is genetic. A mutation in the gene coding for factor VIII (for type A) or IX (for type B) will lead to malfunctions during its synthesis. It will either be completely absent or present in an insufficient quantity, meaning that it won’t have an effective action.
The genetic mutation responsible for hemophilia is recessive and linked to the X chromosome. Since men carry only one of these chromosomes (XY), as opposed to women who carry two (XX), they are therefore much more affected by hemophilia than women. It is very rare for a woman to have hemophilia. As a matter of fact, for a woman to have moderate or severe hemophilia, her father would need to be a hemophiliac and her mother must pass on her X chromosome with the mutation. A woman with only one X chromosome carrying the mutation will not be ill but will carry the disease (functional clotting, or a minor case, but able to pass it on to offspring).
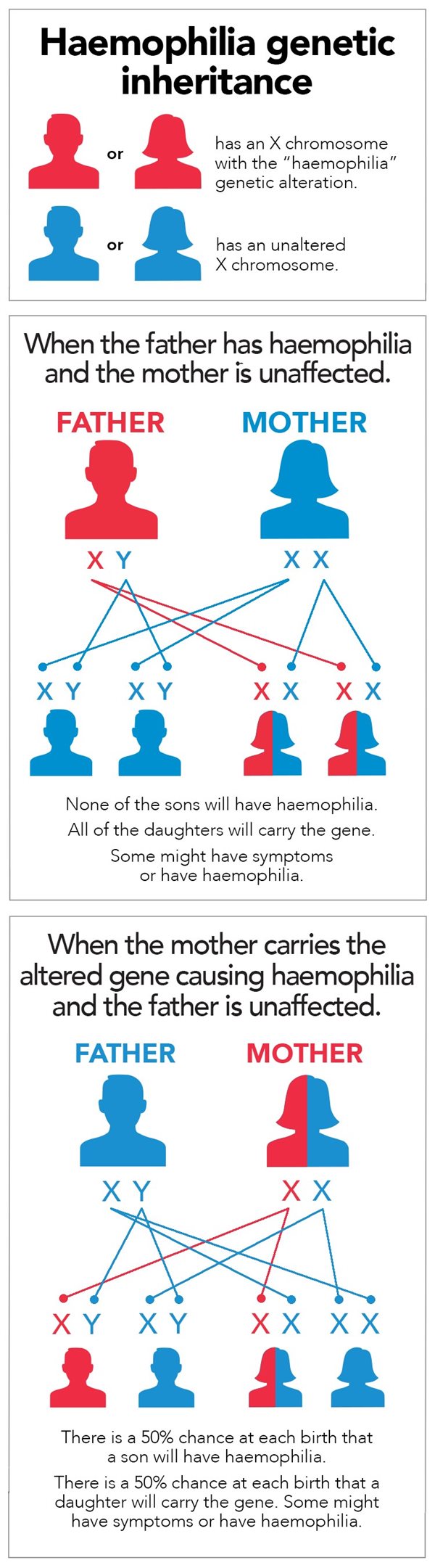
Source: Haemophilia Foundation Australia
In addition, hemophilia A is much more common than B, with one boy in 5,000 births affected by the former, compared to one in 30,000 for the latter.
However, the severity of the hemophilia tends to differ depending on the case. This difference is based on the nature of the mutation. If the mutation causes a complete absence of the clotting factor, the severity will be greater than if the level of the factor is reduced. It is estimated that it is severe in around half of cases, minor in 30-40% of cases and normal in the rest.
There are also rare cases (one in a million per year) of acquired hemophilia, or cases that develop spontaneously later in life. Acquired hemophilia is a type of autoimmune disease where the immune system mistakes the body's clotting factor as a foreign substance and makes antibodies to destroy it. In this type of hemophilia, men and women are equally affected. The origin of the immune dysfunction may be due to an autoimmune condition or a malignant pathology, although it remains unknown in half of the cases.
Diagnosis
Hemophilia is usually diagnosed early (in the first year of life for severe forms), partly because of its hereditary nature and partly because of its characteristic symptoms. For moderate or minor forms, the diagnosis can be made later.
Symptoms:
- Spontaneous bleeding
- Prolonged bleeding after trauma or a surgery
- Bruising
- Hematomas
- Blood in the joints or the muscles
These symptoms are not expressed in all forms of the disease. A mild or minor form will not cause spontaneous bleeding, for example.
If there is a clinical suspicion of hemophilia, it will be necessary to carry out a series of clotting tests. Following a blood test, an increased PTT (partial thromboplastin time) will be the first sign of hemophilia (along with prothrombin time (PT), fibrinogen (factor I) and platelet count all at normal levels). Subsequently, a factor VIII and IX level below 40% will confirm the diagnosis. In the case of suspected hemophilia A, a von Willebrand factor (VWF) test (which protects factor VIII) is also necessary to make the differential diagnosis between von Willebrand disease (VWD) and hemophilia A.
Treatments
The standard treatment for hemophilia is replacement therapy, or supplying the missing clotting factor (factor VIII for type A and factor IX for type B). These factors can be derived from human blood or plasma (from a donor) or from recombinant sources (produced by genetic engineering). The latter have the advantage of limiting the risk of contamination by the donor (with HIV, hepatitis B, etc.).
These substitution treatments can be administered prophylactically (to prevent any bleeding in severe and moderate forms). However, the life span of a clotting factor is relatively short. Thus, the patient will need to receive injections up to several times a week depending on how fast the body consumes them. They can also be taken on demand (for less severe forms or during accidents).
The major problem with replacement clotting factors is the immune system reaction. Indeed, the immune system will develop antibodies directed against these factors. These antibodies, called inhibitors, clump together on their target to inactivate it and attract other immune cells. Higher doses of clotting factor will then be needed to achieve the same clinical response.
Another treatment is also available to low-severity hemophilia A: Desmopressin (DDAVP). This anti-diuretic molecule is also capable of forcing the cells to release the factor VIII and von Willebrand factor they contain. However, its effects may fade with repeated administration over a short period. In addition, DDAVP may not be effective for all patients, so a DDAVP challenge test may be required in order to determine if it is a suitable treatment option for the patient.
Looking forward
Research is now focusing on improving replacement treatments. Several approaches are being studied, including increasing the half-life of clotting factors by binding them to proteins with a long half-life. A trial with Immunoglobulin G (IgG) (the main type of antibody found in the blood) has shown promising results: the half-life was increased three to five times. A longer half-life leads to a longer duration of action.
Additionally, it has been posited that clotting factors without sugars on their surface may be able to limit their recognition by the immune system and thus limit inhibitors.
Finally, gene therapy could directly correct the anomalies. In December 2011, a conclusive trial was conducted in which the gene coding for factor IX (type B) was sent to the liver (its place of synthesis) via an adenovirus. The six patients included in the study were not cured, but the severity of their hemophilia was reduced. It should be noted that this technique would be much more difficult for type A because its gene is much longer.
Was this article helpful to you?
Share your thoughts and questions with the community in the comments below!
Take care!
 Facebook
Facebook Twitter
TwitterSources:
- Comprendre l’hémophilie, Roche
- Hémophilie : définition et symptômes, Roche
- Hémophilie, INSERM
- Qu’est-ce que l’hémophilie acquise ?, Association française des hémophiles
- Protocole national de diagnostic et de soins (PNDS) HEMOPHILIE, HAS
- Hémophilie, Orphanet
- Haemophilia - General Questions, Haemophilia Foundation Australia



